Science Q&A: How SickKids scientists are working to understand a potentially lethal brain disease
Summary:
Dr. Brent Derry, Senior Scientist in the Developmental & Stem Cell Biology Program at SickKids, recently had two studies published on cerebral cavernous malformations, the result of work in his lab at SickKids and in collaboration with colleagues around the world.
Cerebral cavernous malformations (CCM) is a rare disease that causes anomalies in tiny capillaries that transport blood throughout the brain. CCM presents as headaches, seizures, and even hemorrhagic stroke in affected patients. The disease involves defects in one of three CCM genes (CCM1, CCM2, or CCM3). Dr. Brent Derry, Senior Scientist in the Developmental & Stem Cell Biology Program at SickKids, recently had two studies published on CCM, the result of work in his lab at SickKids and in collaboration with colleagues around the world. The first, Interrogating the CCM-3 Gene Network was the cover story in the Sept. 11 issue of Cell Reports. The second, Systemic pharmacological screens uncover novel pathways involved in cerebral cavernous malformations, was also the cover story in the October 9 issue of EMBO Molecular Medicine. We sat down with him to learn more about his research.
What did your research find?
Surgical resection is currently the only curative option for CCM patients, but when lesions are seated deeply in regions of the brain not accessible to neurosurgery, the condition may cause severe illness or death due to recurrent cerebral hemorrhages. It is important to not only understand the functions of the genes mutated in this disease but how they cooperate with other genes in order to develop effective drug therapies. Interventions are desperately needed to (i) prevent the growth and bleeding of existing lesions, and (ii) to suppress the formation of new ones.
The first study in Cell Reports was aimed at defining the network of genes that cooperate with the CCM3 gene, which is associated with the earliest onset and the most aggressive from of the disease. A mutated or lost CCM3 gene leads to cystic capillaries in the brain, so we need to find ways to correct these defects when the gene is absent.
Our team used the excretory system of the nematode worm (C. elegans), which has structural similarities with human brain capillaries affected in CCM patients.
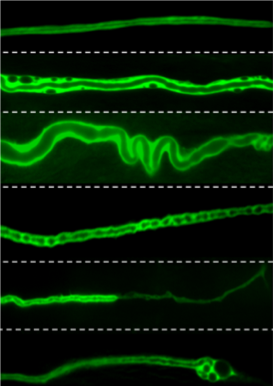
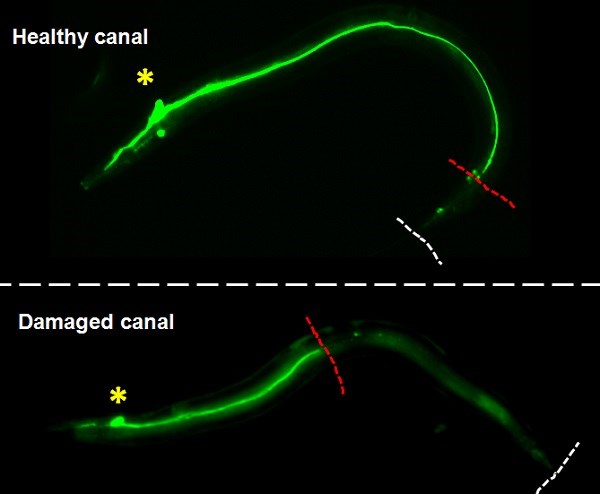
Lead by postdoctoral fellow and first author Dr. Benjamin Lant, our team was the first to perform a genome-wide screen for genes that exhibit similar properties as CCM3, which uncovered over 500 genes including one called MO25. Ablation of MO25 causes the same defects (excretory canal truncations and cysts) as loss of CCM3 in worms. Our collaborators, Dr. Corinne Albiges-Rizo and Dr. Eva Faurobert, showed that depletion of MO25 in human endothelial cells also caused the same defects as depletion of CCM3. This is important because it demonstrates the utility of using the worm to discover genes that exhibit the same functions as their human counterpart, and for the first time implicates MO25 in the pathobiology of CCM. Obviously, much work needs to be done to determine if MO25 is a modifier of CCM disease in patients, and if it might be used therapeutically for treating CCM patients in the future.
The second study in EMBO Molecular Medicine was complimentary to the genetic study, which identified potential therapeutic targets. Here, we focused on identifying small compounds, many of which have been used for treating other diseases in humans, that reverse the defects caused by mutations in CCM genes. This study uncovered several compounds as potential drug therapies that included indirubin-3-monoxime (an active component in traditional Chinese medicine), retinoic acid (a molecule derived from vitamin A) and vitamin D.
This was the most comprehensive screen for CCM active compounds performed to date, and the first demonstration of indirubin-3-monoxime as a potential therapeutic for suppressing the formation of CCM lesions in humans. The findings have profound implications for understanding how the CCM genes function and for treating CCM patients.
What are the next steps for your research?
We need to compare the genes with the most striking similarities to CCM with data from sequencing studies on patients who have the same CCM gene mutation but have a range of severity of symptoms. The main question here is whether the genes we identified in C. elegans have similar functions in CCM patients, like we found for MO25. For example, do mutations in these genes also occur in patients, and if so, do they contribute to the severity of their disease?
For the drug screen, we need to carry out more comprehensive pre-clinical and safety studies to see if these compounds (such as indirubin-3-monoxime, retinoic acid, and vitamin D) can be used in a clinical trial to treat CCM.
Finally, we have an enormous resource to help in the development of CCM therapies. The first study in Cell Reports identified a number of genes that cooperate with CCM3 (and potentially CCM1 and CCM2); we are interested in knowing how they can be targeted by drugs to prevent lesion maturation and/or formation.
Funding for these two projects was provided by the National Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Institutes of Health Research (CIHR) with an international team grant from E-RARE International (with partners in Germany and France).